Epinephrine, but not dexamethasone, induces apoptosis in human retinal pigment epithelium cells in vitro: Possible implications on the pathogenesis of central serous chorioretinopathy
Santiago Antonio B. Sibayan, MD, PhD, Karin Kobuch, MD, Detlev Spiegel, MD, Elfriede Eckert, MTA, Rita Leser, MTA, Jan Monzer, PhD, Veit-Peter Gabel, MD
APOPTOSIS (programmed cell death) is a generegulated process that plays an important role in the normal physiologic turnover of cells and in various pathological processes. It differs from necrosis in a variety of morphologic and biochemical features. Morphologic changes in necrosis include cell swelling, degradation of organelles, rupture of cell membranes, and spillage of cellular contents into the extracellular space, resulting in an inflammatory response.
In contrast, apoptosis is characterized by condensation of the nucleus and cytoplasm, compaction and margination of chromatin, structural disorganization of the nucleus, and formation of cell fragments or “apoptotic bodies”. Apoptotic bodies maintain intact organelles and cell membranes, and are engulfed by neighboring phagocytic cells, thus no inflammatory response is elicited. Biochemical changes in apoptosis are characterized by enzymatic cleavage of DNA by endogenous endonuclease, often into multiples of 180 – 200 base pairs. In necrosis, DNA is cleaved into segments of random length. 6 Apoptosis may occur spontaneously or in response to physiologic stimuli which trigger cellular and molecular transformations that result in cell death. It may also be induced by a variety of exogenous toxic agents. In particular, it has been demonstrated that epinephrine can induce apoptosis in trabecular meshwork cells 7 and thymocytes, 8 and that corticosteroids can cause apoptosis in thymocytes, 5,9,10 eosinophils, 11 and leukemia cells. 12 The retinal pigment epithelium (RPE) performs a variety of functions, which include maintenance of homeostasis of the subretinal space and prevention of free water movement from choroid to retina. Central serous chorioretinopathy (CSCR) is a condition characterized by accumulation of fluid in the subretinal space, resulting in blurring of vision and metamorphopsia. The mechanism by which CSCR is caused is poorly understood. However, it is believed to result from dysfunction of the RPE and/or choroid. CSCR has been noted to be more prevalent among “hard-driving, tense, type-A” individuals who have high levels of endogenous catecholamine (i.e., epinephrine). In experiments performed in monkeys, it has been shown that intravascular administration of epinephrine can induce CSCR, and that the RPE underlying areas of CSCR undergoes degeneration. CSCR has also been noted to develop following systemic, 19,20,21,22,23 epidural 24 and inhalatory or intranasal 25 administration of corticosteroids. Whether apoptosis plays a role in the degeneration of human RPE cells and pathogenesis of CSCR has thus far not been investigated.
The aim of this study is to investigate whether epinephrine and dexamethasone, a corticosteroid, can induce apoptosis in human RPE cells in vitro. These findings may help elucidate the pathophysiologic mechanisms involved in CSCR.
METHODOLOGY
Retinal Pigment Epithelium Cell Cultures
Primary RPE cell cultures were prepared from the eyes of human donors after securing the necessary consent. The donors ranged in age from 28 to 50 years. The cells were processed within 48 hours of death. Methods similar to those described by Sheu et al. 26 were used to harvest the cells. Each eye was incised along the equator, and the anterior segment and vitreous were removed. The retina was carefully peeled off and cut away from the optic disc. The posterior half of the globe was then incised radially and the RPE was mechanically scraped with a cell scraper. The RPE was transferred to culture medium consisting of Dulbecco’s Modified Eagle’s Medium with 10% fetal bovine serum and 1% penicillin-streptomycin (Gibco, Buffalo, NY, USA), and grown to confluence in 25 cm2 tissue culture flasks (Greiner, Frickenhausen, Germany) maintained in an atmosphere of 5% carbon dioxide, 95% relative humidity, at a temperature of 37° C. Upon reaching confluence, the cells were trypsinized and subcultured in 75 cm2 tissue culture flasks (Greiner, Frickenhausen, Germany). All cells used for the experiments were in the third passage and were grown to confluence on 4-well chamber slides (Lab-Tek/ Nalge Nunc, Naperville, IL, USA). All cells in the primar y cell culture had regular polygonal epitheloid shapes and melanin granules, indicating that the cells were of homogenous RPE origin. In addition, the epithelial origin of the cultured RPE cells was confirmed by positive staining with mousederived anti-pancytokeratin Lu-5 (Dianova, Hamburg, Germany). For cytokeratin staining, the following technique was used. RPE cultures were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) for 30 minutes. They were then rinsed in PBS and incubated in 1% bovine serum albumin (BSA) for 10 minutes to block non-specific binding sites. The cultures were subsequently incubated with antipancytokeratin antibody diluted in PBS (working dilution = 1:20) for 60 minutes in a humidified chamber at 37° C. Negative controls were obtained by substituting the antipancytokeratin antibody with PBS. Following incubation, the cultures were washed in PBS and incubated in a 1:100 dilution of fluoresceinconjugated rabbit antimouse antibody (Sigma, St. Louis, MO, USA) at room temperature for 45 minutes in a humidified chamber. The slides were given a final rinse with PBS, coverslipped using the Slow Fade Light Antifade Kit (Molecular Probes, Eugene, OR, USA) as a mounting medium, and viewed with a fluorescence microscope (Leica Model DMRBE, Heerbrugg, Switzerland).
Drug Solutions
RPE cell cultures were incubated in the following drugs (Sigma, St. Louis, MO, USA) dissolved in the previously described culture medium at specified concentration ranges: epinephrine (10 2 – 8×10 7 pg/ml); dexamethasone (4 – 4×10 4 ng/ml). The drug concentrations used in this study are similar to those used in previous experiments involving cultured bovine trabecular meshwork cells. 7 In addition, concentrations of epinephrine and dexamethasone similar to those found in circulating human plasma (epinephrine = 5×10 1 – 5×10 2 pg/ml 27 ; dexamethasone = 3.7 – 13.16 ng/ml following oral administration in normal subjects 28 ) were tested. Cell cultures incubated in drug-free culture medium served as controls. All experiments were performed in quadruplicate.
Morphology Studies
RPE cell cultures were evaluated serially for morphologic changes by phase contrast microscopy with the use of an inverted microscope (Olympus Model CK2, Tokyo, Japan).
In Situ Apoptosis Staining
RPE cells were processed for in situ apoptosis labeling using the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) technique. The In Situ Cell Death Detection Kit (Boehringer Mannheim, Mannheim, Germany) was utilized. This assay is a histochemical stain that labels 3′-OH ends of DNA fragments that are generated during the process of apoptosis. Nonapoptotic cells have relatively insignificant amounts of 3′-OH overhangs, and thus are not stained. The following is a summary of the in situ staining procedure. After incubation with the drugs, the RPE cell cultures were fixed with 4% paraformaldehyde in PBS for 30 minutes. The cells were subsequently washed three times in PBS for 5 minutes each wash. The PBS was aspirated, fluorescein-conjugated TUNEL reaction mixture was applied, and parafilm slips were placed over the specimens to spread the reagent evenly. The specimens were then incubated for 90 minutes in a humidified chamber at a temperature of 37° C. The parafilm slips were removed, the specimens were rinsed three times in PBS, and coverslips were applied on the slides using the Slow Fade Light Antifade Kit as a mounting medium. The slides were then examined with a fluorescence microscope. Positive controls were obtained by incubating nondrugtreated RPE cells in DNAse I (1 mg/ml) (Boehringer Mannheim, Mannheim, Germany) for 10 minutes at room temperature between the fixation and TUNEL reaction steps of the staining procedure. 29 Negative controls were obtained by substituting TUNEL reaction mixture with terminal-transferase-free label solution.
Quantitation of Apoptotic Cells
Quantitation of apoptotic cells was performed by counting the number of apoptotic cells per 200x field. This figure was divided by the total number of cells within the same field, yielding the percentage of apoptotic cells per specified field (the total number of cells per field ranged from 55 – 90 in the epinephrine series and 62 – 92 in the dexamethasone series). Statistical analyses were performed using the Student’s t-test, with a p value less than 0.05 considered statistically significant.
RESULTS
Epinephrine
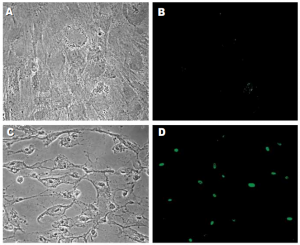
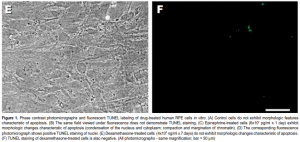
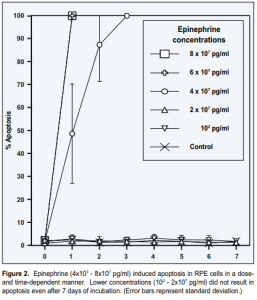
Following treatment with epinephrine (4×10 7 – 8×10 7 pg/ml), RPE cells underwent apoptosis, as evidenced by development of characteristic morphologic changes (condensation of the nucleus and cytoplasm; compaction and margination of chromatin) (Figure 1C) and positive TUNEL staining (Figure 1D). Apoptosis was induced in a dose- and time-dependent manner (Figure 2). Exposure to lower concentrations of epinephrine (10 2 – 2×10 7 pg/ml) for up to 7 days did not result in apoptosis.
Dexamethasone
No morphologic changes consistent with apoptosis were observed in any of the specimens treated with dexamethasone even after 7 days of incubation (Figure 1E). TUNEL staining was also negative in all specimens (Figure 1F).
DISCUSSION
Using a monkey model of epinephrine-induced CSCR, Yoshioka and Katsume 13 have demonstrated degeneration of the RPE underlying areas of CSCR. Our finding that epinephrine can induce apoptosis in RPE cells may help substantiate this mechanism of degeneration and may contribute to the explanation of the pathogenesis of CSCR. It has previously been shown that stimulation of betaadrenergic receptors in RPE cells in vitro causes a net increase in cyclic adenosine 3′, 5′-monophosphate (cAMP) levels. 30 It has also been demonstrated that agents that elevate cAMP levels (i.e., epinephrine) can induce apoptosis. 31 These findings suggest that apoptosis in RPE cells may be induced by a beta-adrenergic/cAMP-mediated process. Clinical observations suggest that systemic administration of beta-blockers to patients with CSCR may improve vision and hinder recurrence of CSCR. 32 If RPE cell apoptosis indeed occurs in CSCR, these observations further support the hypothesis that it is mediated by a beta-adrenergic mechanism. Another possible mechanism by which apoptosis could be induced may involve oxidative byproducts of epinephrine. Epinephrine is metabolized by monoamine oxidase (MAO) into 3,4-dihydroxyphenylglycolaldehyde (DOPEGAL) and hydrogen peroxide, 33 both of which could act as inducers of apoptosis. 33,34,35 Experimental evidence suggests that MAO is present in RPE cells. 36 RPE cells could therefore metabolize epinephrine and undergo apoptosis in response to its metabolites. Drug doses used in toxicity studies include high, intermediate and low levels of a test compound. High doses allow one to observe toxic effects more readily. 37 In the current study, epinephrine levels at which apoptosis was observed were 8 x 10 4 to 1.6 x 10 6 times higher than endogenous in vivo levels. Since apoptosis could be difficult to detect due to its rapid kinetics and the relative paucity of cells that undergo apoptosis at any specific time point, 38 the administration of such high drug doses could facilitate observation of this process. Further toxicokinetic studies would however be necessary to determine how these high-dose acute findings correlate with the in vivo situation where prolonged exposure to lower drug levels is present. 37 Dexamethasone did not induce apoptosis at any of the tested concentrations. However, corticosteroids may cause an upregulation of adrenergic response by increasing the number of adrenergic receptors and/or increasing the responsiveness of each receptor. 39 A corticosteroidinduced increase in adrenergic response could possibly result in a cAMP-mediated apoptotic response similar to that observed in epinephrine-treated cells, and could also account for CSCR observed following treatment with corticosteroids. Such responses however could not be evaluated in these studies since no adrenergic agents were present in the cell cultures treated with dexamethasone. Although the pathophysiology of CSCR is poorly understood, various theories regarding its pathogenesis have been proposed. Spitznas 14 hypothesized that it is due to a cAMP-mediated reversal of the RPE fluid pumping action. According to this model, fluid is pumped in a chorioretinal, instead of a retinochoroidal direction. This leads to accumulation of fluid in the subretinal space. Initially, the fluid movement occurs transcellularly. However, the diffusion barrier (i.e., RPE) eventually breaks down, leading to direct leakage of fluid into the subretinal space. Although Spitznas proposed that the disruption of the barrier is due to strong fluid flow, it could also be due to degeneration of the RPE. Applying our findings to this model, the increase in cAMP could account not only for the reversal of the pumping action, but also for the breakdown of the diffusion barrier.
Gass 15 suggested that CSCR is due to focal areas of increased permeability in the choriocapillaris. Yoshioka and Katsume 13 and Marmor 16,17 proposed that defects in both the choroid and RPE are necessary for leakage of fluid into the subretinal space. The latter proposal is supported by the experimental observation that, in addition to degeneration of the RPE, diaphragms of the fenestrated endothelial cells on the inner sur face of the choriocapillaris disappear. 13 These defects in the choroid and RPE create a pressure head, resulting in such leakage. While our findings cannot fully explain the observed anatomic changes and proposed pathophysiologic mechanisms involved in CSCR, they could contribute to the explanation of the RPE component of this condition. Whether epinephrine causes CSCR by inducing apoptosis in RPE cells in vivo cannot be determined from this study. However, our findings provide a possible pathophysiologic mechanism for this poorly understood disease entity.
2. Kerr JFR, Searle J, Harmon BV, Bishop CJ. Apoptosis. In: Potten CS, ed. Perspectives on mammalian cell death. New York: Oxford University Press; 1987: 93-128.
3. Schwartzman RA, Cidlowski JA. Apoptosis: The biochemistry and molecular biology of programmed cell death. Endocrine Rev. 1993; 14: 133-151.
4. Kerr JFR, Wyllie AH, Currie AR. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. 1972; 26: 239-257.
5. Wyllie AH. Glucocorticoid-induced thymocyte apoptosis is associated with endogenous endonuclease activation. Nature. 1980; 284: 555-556.
6. Afanas’ev VN, Korol’ BA, Mantsygin YA, Nelipovich PA, Pechatnikov VA, Umansky SR. Flow cytometry and biochemical analysis of DNA degradation characteristic of two types of cell death. FEBS Lett. 1986; 194: 347-350.
7. Sibayan SAB, Latina MA, Sherwood ME, Flotte TJ, White K. Apoptosis and morphologic changes in trabecular meshwork cells in vitro. Exp Eye Res. 1998; 66: 521-529.
8. Rinner I, Kukulansky T, Skreiner E, et al. Adrenergic and cholinergic regulation of apoptosis and differentiation of thymic lymphocytes. Adv Exp Med Biol. 1994; 355: 113-117.
9. Cohen JJ, Duke RC. Glucocorticoid activation of a calcium-dependent endonuclease in thymocyte nuclei leads to cell death. J Immunol. 1984; 132: 38-42.
10. Compton MM, Cidlowski JA. Rapid in vivo effects of glucocorticoids on the integrity of rat lymphocyte genomic deoxyribonucleic acid. Endocrinology. 1986; 118: 38-45.
11. Kawabori S, Soda K, Perdue MH, Bienenstock J. The dynamics of intestinal eosinophil depletion in rats treated with dexamethasone. Lab Invest. 1991; 64: 224-233.
12. McConkey DJ, Aguilar-Santelises M, Hartzell P, et al. Induction of DNA fragmentation in chronic B-lymphocytic leukemia cells. J Immunol. 1991; 146: 1072-1076.
13. Yoshioka H, Katsume Y. Experimental central serous chorioretinopathy. III: Ultrastructural findings. Jpn J Ophthalmol. 1982; 26: 397-409.
14. Spitznas M. Pathogenesis of central serous retinopathy: A new working hypothesis. Graefe’s Arch Clin Exp Ophthalmol. 1986; 224: 321-324.
15. Gass JDM. Pathogenesis of disciform detachment of the neuroepithelium. II: Idiopathic central serous choiroidopathy. Am J Ophthalmol. 1967; 63: 587-615.
16. Marmor MF. New hypotheses on the pathogenesis and treatment of serous retinal detachment. Graefe’s Arch Clin Exp Ophthalmol. 1988; 226: 548-552.
17. Marmor MF. On the cause of serous detachments and acute central serous chorioretinopathy. Br J Ophthalmol. 1997; 81: 812-813.
18. Yanuzzi LA. Type A behavior and central serous chorioretinopathy. Trans Am Ophthalmol Soc. 1986; 84: 799-845.
19. Jain IS, Singh K. Maculopathy a corticosteroid side-effect. J All India Ophthalmol Soc. 1966; 14: 250-252.
20. Polak BCP, Seerp Baarsma G, Snyers B. Diffuse retinal pigment epitheliopathy complicating systemic corticosteroid treatment. Br J Ophthalmol. 1995; 79: 922-925.
21. Spraul CW, Lang GE, Lang GK. Retinal pigment epithelial changes associated with systemic corticosteroid treatment: Report of cases and review of the literature. Ophthalmologica. 1998; 212: 142-148.
22. Hooymans JMM. Fibrotic scar formation in central serous chorioretinopathy developed during systemic treatment with corticosteroids. Graefe’s Arch Exp Clin Ophthalmol. 1998; 236: 876-879.
23. Bouzas EA, Moret P, Pournaras CJ. Central serous chorioretinopathy complicating solar retinopathy treated with glucocorticoids. Graefe’s Arch Clin Exp Ophthalmol. 1999; 237: 166-168.
24. Kao LY. Bilateral serous retinal detachment resembling central serous chorioretinopathy following epidural steroid injection. Retina. 1998; 18: 479-481.
25. Haimovici R, Gragoudas ES, Duker JS, Sjaarda RN, Eliott D. Central serous chorioretinopathy associated with inhaled or intranasal corticosteroids. Ophthalmology. 1997; 104: 1653-1660.
26. Sheu SJ, Sakamoto T, Osusky R, et al. Transforming growth factor-ß regulates human retinal pigment epithelial phagocytosis by influencing a protein kinase Cdependent pathway. Graefe’s Arch Clin Exp Ophthalmol. 1994; 232: 695-701.
27. O’Connor DT. Adrenal medulla. In: Bennett JC, Plum F, eds. Cecil Textbook of Medicine. Philadelphia: W.B. Saunders; 1996: 1253-1257.
28. Meikle AW, Lagerquist LG, Tyler FH. Apparently normal pituitary-adrenal suppressibility in Cushing’s syndrome: Dexamethasone metabolism and plasma levels. J Lab Clin Med. 1975; 86: 472-478.
29. Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J Cell Biol. 1992; 119: 493-501.
30. Frambach DA, Fain GL, Farber DB, Bok D. Beta adrenergic receptors on cultured human retinal pigment epithelium. Invest Ophthalmol Vis Sci. 1990; 31: 1767-1772.
31. McConkey DJ, Orrenius S, Jondal M. Agents that elevate cAMP stimulate DNA fragmentation in thymocytes. J Immunol. 1990; 145: 1227-1230.
32. Avci R, Deutman AF. Die Behandlung der zentralen serösen Choroidopathie mit dem Betarezeptorenblocker Metoprolol (vorläufige Ergebnisse). Klin Monatsbl Augenheilkd. 1993; 202: 199-205.
33. Burke WJ, Kristal BS, Yu BP, Li SW, Lin TS. Norepinephrine transmtter metabolite generates free radicals and activates mitochondrial permeability transition: A mechanism for DOPEGAL-induced apoptosis. Brain Res. 1998; 787: 328-332.
34. Burke WJ, Schmitt CA, Miller C, Li SW. Norepinephrine transmitter metabolite induces apoptosis in differentiated rat pheochromocytoma cells. Brain Res. 1997; 760: 290-293.
35. Li PF, Dietz R, von Harsdorf R. Reactive oxygen species induce apoptosis of vascular smooth muscle cell. FEBS Lett. 1997; 404: 249-252.
36. Moroi-Fetters SE, Earley O, Hirakata A, Caron MG, Jaffee GJ. Binding, coupling and mRNA subtype heterogeneity of α1-adrenergic receptors in cultured human RPE. Exp Eye Res. 1995; 60: 527-532.
37. Chasseaud LF. Role of toxicokinetics in toxicity testing. In: Welling PG, de la Iglesia FA, eds. Drug toxicokinetics. New York: Marcel Dekker Inc.; 1993: 105-142.
38. Coles HSR, Burne JF, Raff MC. Large-scale normal cell death in the developing rat kidney and its reduction by epidermal growth factor. Development. 1993; 118: 777-784.
39. Davies AO. Steroid hormone-induced regulation of adrenergic receptors. In: Schleimer RP, Claman HN, Oronsky AL, eds. Anti-inflammatory steroid action: Basic and clinical aspects. San Diego: Academic Press; 1989: 96-109.
Acknowledgments
The authors wish to acknowledge the following individuals for their generous assistance: Dr. Manuel Agulto, Dr. Mario Aquino, Mr. Markus Bock, Mrs. Susan M. Sibayan, Mrs. Brigitte Eßer-Trojan, Dr. Thomas J. Flotte, Dr. Mohandas Kini, Dr. Jaime V. Lapus, Dr. Mark A. Latina, Dr. Juan R. Nañagas, Dr. Markus Preising, Dr. Salvador Salceda, Dr. Prospero Tuaño and Dr. Kristin White.