Juvenile Onset Open-Angle Glaucoma in a Large Family in Cebu, Philippines
Justine May R. Torregosa, MD, DBPO
Christine Siguan-Bell MD, DPBO
Cebu Institute of Medicine, Cebu Velez General Hospital, Cebu
Corresponding Author: Justine May Torregosa, MD
Clinic Address: ACES Eye Referral Clinics, 10th Floor Velez Medical Arts Bldg, V. Ranudo Street, Cebu City, Philippines, 6000
Contact Number: (032) 254-8691 or +639957192563
Email Address: jm.torregosa@gmail.com
Disclosure: The authors report no conflict of interests.
INTRODUCTION
Glaucoma refers to a group of diseases characterized by optic neuropathy that is consistent with neural and connective tissue damage and corresponding patterns of visual field loss.1 JOAG is a rare form of primary open-angle glaucoma (POAG) that presents between ages 3 and 35 years old.1,2 Ocular hypertension, on the other hand, is defined as IOPs of more than 21 mmHg, but with normal optic disc, retinal nerve fiber layer, visual fields, and open angles.2
A family history of glaucoma has long been recognized as a major risk factor for the disease. JOAG is usually inherited as a Mendelian trait with high penetrance whereas the adult-onset POAG has a more complex trait inheritance.3,4 Patients with JOAG show male preponderance, myopia, and elevated IOPs. Gonioscopic findings show open angle with variable degree of goniodysgenesis. Bilateral involvement is often present in those with positive family history of glaucoma. Progressive visual field loss is also more likely with higher baseline IOPs.5,6
The exact mechanism of glaucoma damage in open-angle glaucoma is unknown. Studies done on large families with JOAG have led to the discovery of a genetic loci called GLC1, wherein the MYOC gene that codes for the myocilin protein has been identified. Mutation is said to result in reduced solubility of the intracellular protein, leading to protein misfolding, subsequent protein aggregation in the endoplasmic reticulum, and eventual loss of trabecular meshwork cells.4
Future directions in glaucoma studies are looking into genotype-phenotype correlations and clinical outcome studies. As more genes are being identified by wide-based genetic mapping, clinical features that define the glaucoma phenotype are also being recorded.4
This study documented the clinical profile of a four-generation Cebuano family with JOAG.
METHODOLOGY
This study was conducted at the outpatient department of Cebu Velez General Hospital after obtaining approval from its institutional review board and carried out in accordance with the tenets of the Declaration of Helsinki. Thirty-eight (38) members of a four-generation family from the northern part of Cebu province were examined. General medical and ophthalmic histories were taken. Ophthalmologic examination included visual acuity measurements, refraction, slit-lamp examination, IOP measurements, gonioscopy, optic nerve evaluation, and standard automated perimetry (SAP). Individuals were considered to have JOAG if they have the following characteristics: (1) onset of symptoms between 4 and 40 years of age; (2) absence of secondary causes of glaucoma; (3) IOPs of more than 21 mmHg in at least one eye; (4) open anterior chamber angles [grade 3 or 4 on modified Shaeffer’s classification]; and (5) characteristic optic nerve damage and/or visual field loss typical of glaucoma. Family members who had elevated IOP but did not have the characteristic optic nerve damage and/or visual field loss were classified under ocular hypertension. Asymptomatic family members were also invited to participate and undergo clinical evaluation.
There were 7 family members, including the proband, who had records of their previous examination and treatment in our institution; these were obtained and analyzed. They underwent SAP using Octopus 600 Perimeter (Haag-Streit, USA). Severity of the field defects were classified using Hodapp-Parrish-Anderson Classification.2
Clinical examination findings were recorded and tabulated using Microsoft Excel 2011.
RESULTS
The proband is a 31-year-old male diagnosed with JOAG. He came in for glaucoma screening at age 28 years due to his family history. He was initially diagnosed with ocular hypertension with IOPs ranging from 32 to 38 mmHg. He had a normal visual field exam and a non-glaucomatous looking nerve. Anterior segment examination was normal for both eyes (Figure 1A). Gonioscopy showed ciliary body band (CBB) on all four quadrants with flat iris plane and 1+ trabecular meshwork pigmentation (Figure 1B). A monotherapy of prostaglandin analogue was started lowering the IOPs to mid-20s and subsequently shifted to combination topical drugs consisting of prostaglandin analogue with beta-blocker and carbonic anhydrase inhibitor with alpha agonist. After two years of treatment with good compliance, both eyes showed poor responses with IOPs ranging from 28 to 40 mmHg. Repeat visual field exam still revealed no glaucomatous defects but there were emerging glaucomatous changes in the optic nerve head of the right eye (Figure 1C). That eye subsequently underwent trabeculectomy.

The pedigree in this study comprised four generations and showed an autosomal dominant pattern of inheritance (Figure 2). In the first generation, the paternal grandmother was diagnosed with glaucoma. In the second generation, 4 out of 5 siblings (80%) were confirmed cases of glaucoma, including the proband’s father. The third generation had 40 to 75% of offsprings per parent with positive clinical manifestations, with a total of 11 family members diagnosed with JOAG and 1 with ocular hypertension. At the time of the study, the fourth generation, in the age range of newborn to 22 years of age with the majority in the pre-adolescent age, had two family members with glaucoma.
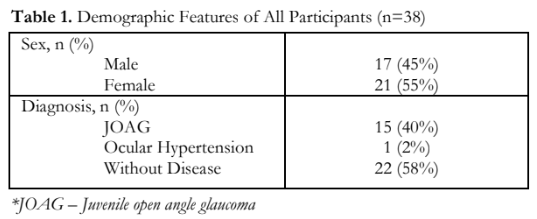
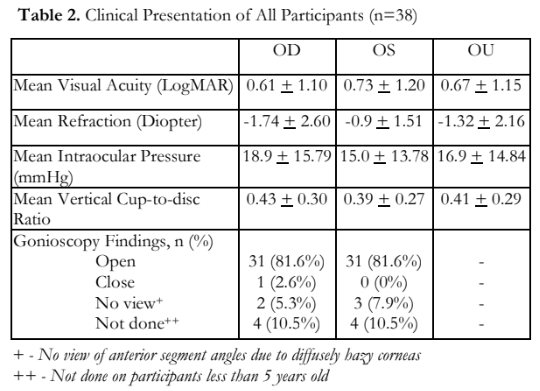
The demographic profile of all the family members who participated in the study are shown in Table 1. Thirty-eight (38) members were included, 17 males and 21 females. Sixteen (42%) had elevated IOPs; 15 (40%), including the proband, were diagnosed with JOAG. One (2%) had ocular hypertension, and 22 (58%) did not have apparent glaucoma. Mean visual acuity was LogMAR 0.67 ±1.15 with a mean refraction of -1.32 + 2.16 diopters. Mean IOP of both eyes was 16.9 + 14.84 mmHg; the mean for the right eye was slightly higher (18.9 +15.79 mmHg) than the left eye (15.02 + 13.78 mmHg). Mean vertical cup-to-disc ratio for all individual family members was 0.41 + 0.29 for both eyes. Gonioscopy using the Posner gonioprism showed almost all members (81.6%) with open anterior chamber angles and flat iris plane. Four members under five years of age were not given consent by their parents to undergo the procedure. Three of the examined individuals had hazy corneas. These individuals have been diagnosed with JOAG in the past, and either underwent surgical treatments with complicated post-surgical courses or were unable to comply with any of the management (Table 2).
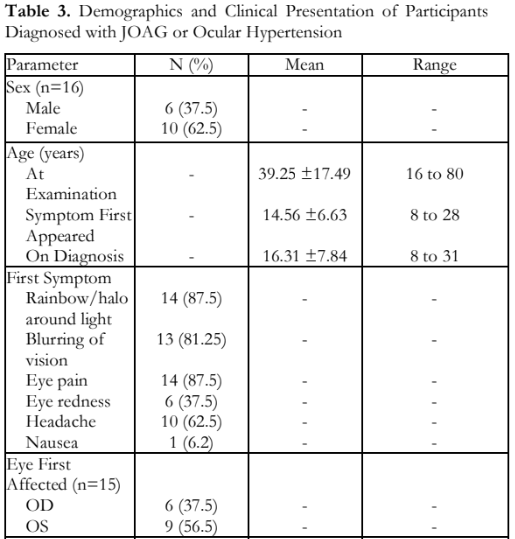
The demographics and clinical presentation of all affected participants are shown in Table 3. Among the 16 patients with JOAG, 6 (37.5%) were males and 10 (62.5%) females, with a mean age at diagnosis of 16.31 + 7.84 years compared to mean age of 14.56 + 6.63 years when symptoms were first noted. One member of the fourth generation first noticed symptoms at 8 years prompting a consultation and was the youngest recorded age at diagnosis (Table 3).
The initial symptoms included the following: rainbow or halos around light (87.5%), blurring of vision and eye pain (81.25%), headaches (62.5%), eye redness (37.5%), and nausea (6.25%). The left eye was affected first in 9 out of 15 participants (56.5%) with glaucoma (Table 3).

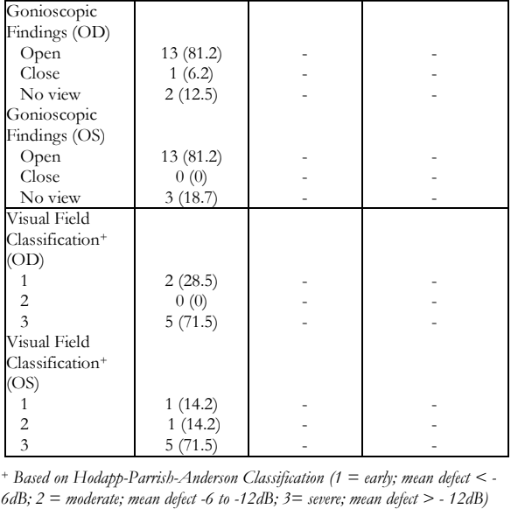
Among affected participants, the mean visual acuity for both eyes was LogMAR 1.54 + 1.34, with a mean refraction of -3.09 + 2.54 diopters. Eyes with high myopia were noted to be buphthalmic. Mean IOP on both eyes was 24.56 + 20.17 mmHg (range: 0 to 65mmHg). IOPs of 0 were recorded in phthisical eyes. Mean vertical cup-to-disc ratio of both eyes was 0.72 + 0.33 (range: 0.3 to 1.0). Gonioscopy revealed open angles with flat iris plane in 81.25% of the examined individuals; only one eye had closed angles which previously were open prior to a complicated post-surgical course. Five out of 7 (71.5%) participants had at least one eye with severe visual field defect (>12dB) (Table 4).

Table 4 shows a summary of treatment received by the participants for JOAG. All participants with elevated IOPs were given topical anti-glaucoma medications, initially started on monotherapy followed by combination drugs. Eleven out of the 15 needed lasers or surgeries: 4 participants had undergone transscleral cyclophotocoagulation, 5 had trabeculectomy in both eyes. One participant had trabeculectomy in one eye and glaucoma drainage device (Ahmed Valve) implantation in the fellow eye.
DISCUSSION
This study described a four-generation family with a strong history of juvenile onset open-angle glaucoma with an autosomal dominant pattern of inheritance. The mean age of onset is 14.56 + 6.63 years, ranging from 8 to 28 years. Mean age at diagnosis in several studies ranged from 18 to 30 years.5-9
IOP is a significant prognostic factor in the progression of the disease. A retrospective study by Kwun concluded that visual field progression was associated with higher IOPs from the latest visit.5 In this study, mean IOP of both eyes at examination was 24.56 + 20.17 mmHg; highest recorded IOP in one eye was 65 mmHg.
Different case series and retrospective studies have demonstrated that low-grade myopia is associated with the development of JOAG and as the disease progresses, the degree of myopia also increases.5,9 The affected participants in this study had a mean refraction of -3.09 + 2.54 diopters.
While there have been variable results with regard to gender predilection among case series published, a retrospective study that included 125 eyes showed male predominance.5,7,9 In this study, there were more female members who had the disease (62.5%).
JOAG has an insidious progression and most of the cases were diagnosed late.1,5,10 Many affected individuals in the first generation had either one or both eyes at the end-stage of glaucoma with blurring of vision the earliest and most common symptom.1,9 This study demonstrated that most of the affected members (87.5%) reported rainbows or halos around light as the first symptom noticed.
Gonioscopy on JOAG patients usually presents as normal.1 In this study, most had open anterior chamber angles with flat iris plane, just like the unaffected members. None showed signs of goniodysgenesis.
The characteristic pattern of disease progression in JOAG is aggressive and generally refractory to medical treatment.1,10 The proband in this study demonstrated the typical disease progression occurring in the family; initially presenting as ocular hypertension with IOP ranging from 30 to 40 mmHg, started on monotherapy and subsequently shifted to combination therapies, followed by the need for surgical interventions.
JOAG tends to follow a characteristic pattern in its clinical presentation, with a positive family history as a major risk factor. To the authors’ knowledge, this is the first case series that described the clinical presentation of JOAG in a large family in the region.
The family in this study demonstrated the typical JOAG phenotype that included early age of onset, strong family history with an autosomal dominant pattern of inheritance in the pedigree, myopia, open angles, and increased IOPs that were refractory to medical treatment.
REFERENCES
- American Academy of Ophthalmology. Basic and clinical science course, 2018-2019. Section 10: Glaucoma. San Francisco: AAO, 2018-2019.
- Aquino MV, Tumbocon JA, De Guzman MHPU. Glaucoma Handbook. Manila: St. Lukes Medical Center – International Eye Institute, 2012.
- Wiggs JL, Damji KF, Haines JL, et al. The distinction between juvenile and adult-onset primary open-angle glaucoma. Am J Hum Genet. 1996;58(1):243-4.
- Wiggs JL. Genetic etiologies of glaucoma. Arch Ophthalmol. 2007;125(1):30-7.
- Kwun Y, Lee EJ, Han JC, Kee C. Clinical Characteristics of Juvenile-onset Open Angle Glaucoma. Korean J Ophthalmol. 2016;30(2):127-33.
- Johnson AT, Richards JE, Boehnke M, et al. Clinical phenotype of juvenile-onset primary open-angle glaucoma linked to chromosome 1q. Ophthalmology. 1996;103(5):808- 14.
- Zhuo YH, Wei YT, Bai YJ, et al. Pro370Leu MYOC gene mutation in a large Chinese family with juvenile-onset open angle glaucoma: correlation between genotype and phenotype. Mol Vis. 2008;22;14:1533-9.
- Zhao X, Yang C, Tong Y, et al. Identification a novel MYOC gene mutation in a Chinese family with juvenile- onset open angle glaucoma. Mol Vis. 2010;16:1728-35.
- Urbak SF. Clinical phenotype of juvenile open-angle glaucoma linked to chromosome 1q in a Danish family. Acta Ophthalmol Scand. 1999;77(5):568-74.
- Noman SM. Juvenile open angle glaucoma –A case series study. Indian J Clin Exp Ophthalmol 2021;7(2):448-451