Manifestations of Bardet-Biedl syndrome
Kristine T. Lo, MD, Juancho Remulla, MD, Alvina Pauline Santiago, MD
BARDET-BIEDL syndrome (BBS) is an autosomal recessive condition characterized by rod-cone dystrophy,
postaxial polydactyly, central obesity, mental retardation, hypogonadism, and renal dysfunction. Other features, though not always present, are hepatic fibrosis, diabetes mellitus, reproductive abnormalities, endocrine abnormalities, short stature, developmental delay, and speech deficits.1 BBS is genetically heterogenous with at least 6 loci mapped to date. The most common gene is BBS1 located at chromosome 11.2 Other genes identified are BBS2 at chromosome 16, BBS3 at chromosome 3, BBS4 at chromosome 15, BBS5 at chromosome 2, and BBS6 at chromosome 20.3 The syndrome is relatively rare. It is estimated to be 1 in 160,000 in Switzerland. It is more common among Arab populations, specifically the Bedouins of Kuwait.4 where interfamilial marriages are common. In Newfoundland, Canada, the incidence is 1 in 17,500. In the Philippines, no case has been reported yet. In this article we describe a case of Bardet-Biedl Syndrome presenting the features seen in the patient.
CASE HISTORY
A 7-year-old boy presented with a history of blurring of vision of two years duration, developmental delay, and
nightblindness. Weighing 5.8 pounds at birth, he was born full term via spontaneous vaginal delivery to a 28-
year-old primigravida, who was on rifampicin for pulmonary tuberculosis during the first 3 months of
pregnancy. The patient was noted to have six digits on all extremities and a small penis. At 3 months of age, he was treated with rifampicin for primary complex. The eldest of 2 children (the sibling is normal), he
was noted to have developmental delay upon consultation, with a developmental age equivalent to a 3- to
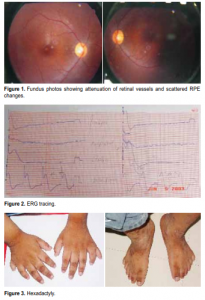
4- year-old. Best-corrected visual acuity was 20/70(6/21) on the right eye and 20/50 (6/15) on the left. Refraction was plano ≈ –4.00 cyl at axis 180 for both eyes. External adnexae were normal. Pupils were 3 mm and briskly reactive to light with no afferent pupillary defect (APD). The patient was orthophoric with a full range of ocular movements. Intraocular pressures were normal. Biomicroscopic examination revealed normal anterior segment. Indirect ophthalmoscopy revealed attenuated retinal vessels, atrophic retinal pigment epithelial (RPE) changes (Figure 1), and sparse pigmentation at the retinal periphery. Electroretinogram (ERG) showed findings consistent with tapetoretinal degeneration and retinitis pigmentosa (Figure 2). Visual-field examination was unreliable. Physical examination showed the patient was obese and had hexadactyly (Figure 3) and micropenis (Figure 4) measuring approximately 2 centimeters in length, with descended testes and a labia-like scrotum. The patient also had small dental roots and hypodontia (Figure 5).
DISCUSSION
Differential diagnoses
Based on the ocular findings and systemic manifestations, the following differential diagnoses were
considered: Weiss syndrome, Biemond II syndrome, Laurence Moon syndrome, Alstrom syndrome and BardetBiedl syndrome. Alstrom syndrome is characterized by tapetoretinal degeneration, obesity, preaxial polydactyly, diabetes mellitus, and neurogenic deafness.5 The patient had only two (obesity, polydactyly) of these manifestations. His blood glucose was normal and he had no symptoms of diabetes. Neither did he have neurogenic deafness. The other findings seen in this patient (hypogenitalism, post axial polydactyly, and learning disability) are not manifestations of Alstrom syndrome. Biemond II syndrome is characterized by ocular defects (more specifically iris coloboma), learning disability, polydactyly, obesity, and hypogenitalism.6
Although the patient had four of the main characteristics, retinal dystrophy is not a characteristic of this syndrome. Obesity, mental deficits, genital dystrophy, ner ve deafness, and short stature characterize Weiss syndrome,6 which were all seen in the patient. But the patient had retinal dystrophy, which is not characteristic of this syndrome. Laurence Moon syndrome is characterized by retinitis pigmentosa or rod cone dystrophy, mental deficiency, hypogenitalism, and spastic paraparesis.6 The patient, however, had no spastic paraparesis and patients with Laurence Moon syndrome do not show polydactyly and obesity. Similar to Laurence Moon syndrome, Bardet-Biedl syndrome is characterized by retinitis pigmentosa, obesity, postaxial polydactyly, learning disabilities, hypogenitalism, and in some cases, renal dysfunction.6 With 5 of the 6 characteristics present in this patient, he was diagnosed to have Bardet-Biedl syndrome.
Diagnostic criteria for Bardet-Biedl syndrome
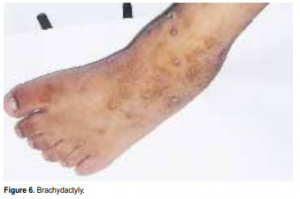
Beales et al. developed the diagnostic criteria for BardetBiedl syndrome: the presence of four primary features or a combination of three primary features plus two secondary features.1 The primary features are rod-cone dystrophy, polydactyly, obesity, learning disabilities, hypogenitalism in males, and renal anomalies. The patient met 5 of these criteria. Secondary features are speech disorder/delay, strabismus, cataracts, astigmatism, syndactyly, brachydactyly, developmental delay, polyuria and polydipsia. The patient had astigmatism, speech disorder, developmental delay, and brachydactyly (Figure 6).

Additional secondary features are hypodontia/small tooth roots, dental crowding, high arched palate, mild spasticity, diabetes mellitus, left ventricular hypertrophy/ congenital heart disease, hepatic fibrosis, ataxia, poor coordination, and imbalance. The patient had hypodontia and small tooth roots. On the average, Bardet-Biedl syndrome is diagnosed at 9 years of age. The relatively late age of diagnosis can be explained by the slow development of clinical features. The first visual disturbance is usually night blindness.
Retinopathy
Retinitis pigmentosa is a disease characterized by progressive photoreceptor degeneration with RPE atrophy
and intraretinal pigment migration of the RPE.7
Symptoms
include progressive contraction of the visual fields and night blindness. Decreased visual acuity can result from macular involvement of the disease. Attenuated vessels, mottling, granularity of the RPE, bone spicule formation, and waxy pale appearance of the disc are typical fundus findings in retinitis pigmentosa. Rod-cone dystrophy found in Bardet-Biedl syndrome is an atypical form of retinitis pigmentosa characterized by early decrease in visual acuity related to early macular involvement. The retinal periphery often has sparse bone spicule pigmentation, with central and peripheral RPE atrophy. Vessel attenuation and disc pallor are also present.8 The retinal findings in this patient were consistent with an atypical presentation. Other findings were constricted visual fields, severe color deficiency, raised dark-adapted thresholds, extinguished and minimal rod and cone responses on ERG. Visual-field testing and color testing were unreliable because the patient was uncooperative. The ERG showed findings consistent with retinitis pigmentosa.
Learning disabilities
Sixty-two percent of Bardet-Biedl syndrome patients have learning disabilities and short-term memory deficit,
which is often counterbalanced by excellent long-term memor y. These patients per form much better at performance rather than verbal skills.1 A recent study in the United Kingdom showed that the majority of children with Bardet-Biedl syndrome were able to remain in mainstream education in the early years. In time, however, almost half required placement in special schools.
Obesity
Obesity is a major problem in this syndrome and is one of the most frequent reasons for medical consultation. It is present in 75% of patients. Excess weight gain does not usually begin until 1-2 years of age. The cause is unknown but the mechanism of obesity appears to be a complex combination of hyperphagia and altered disposal of calories.1
Postaxial polydactyly
The presence of extra digits is a distinct feature of Bardet-Biedl syndrome where 66% of patients have
postaxial hexadactyly ranging from a single skin tag to a fully formed digit on all four limbs. Brachydactyly—having short stubby fingers and toes—is common. When present, it can considerably affect dexterity. Syndactyly is less frequently seen but is usually partial and confined to the feet.1
Hypogenitalism
Hypogenitalism occurs mostly in men; 88% were found to have small testes and very small penises. These patients, however, have normal testosterone levels. Women with Bardet-Biedl syndrome usually have normal secondary characteristics but often have menstrual irregularities and endocrine dysfunction with increased luteinizing hormone.9 This patient had a penis about 2 centimeters in length.
Renal dysfunction
Renal abnormalities can lead to life-threatening problems. Some patients have structural abnormalities in
the form of dysplastic kidneys with irregular contour and cystic dilatation of collecting ducts and calyces. About 15% develop symptomatic renal impairment, with 5% going into end-stage renal failure.10 This patient had normal kidneys on ultrasonography and normal parameters of renal function. Intravenous pyelography may be a useful test in some patients.
Secondary features
The secondary features present in this patient were astigmatism, brachydactyly or short stubby feet, developmental delay, speech disorders and dental problems. This patient did not manifest any cardiac, hepatic or endocrine abnormalities.
Management
Bardet-Biedl syndrome is recessively inherited with both parents being phenotypically normal. The risk of
subsequent offspring being affected is 25%. There is a two-thirds chance that unaffected siblings will be carriers. When a new case is seen, a careful family history should be taken and other relatives examined. In this case, the patient was the only one affected in his family. Beales and coauthors recommend that all patients
suspected of Bardet-Biedl syndrome undergo baseline ERG, renal ultrasound, intravenous pyelography, ECG, and echocardiogram.1 Follow-up include biannual urinalysis and annual blood pressure, serum urea, and
creatinine screening to monitor for development of renal dysfunction. Much can be done to address the ophthalmologic problems of patients with Bardet-Biedl syndrome. Careful refraction should be performed for visual rehabilitation. The patient should also be prepared for a life with low vision. For retinitis pigmentosa, Vitamin A has been found effective in slowing down the decline in ERG responses.7 However, these findings were based on patients with typical retinitis pigmentosa and not syndromic retinitis pigmentosa. Although ocular manifestations may be the most significant aspect of the disease, other systemic problems should be addressed. Treatment of Bardet-Biedl syndrome is directed toward specific organs and systems. A multidisciplinary approach is suggested with the ophthalmologist addressing the problems of retinitis pigmentosa (RP) and visual rehabilitation. The pediatrician should be involved in the overall care of the patient and refer the patient to appropriate subspecialties when necessary. Intervention for renal dysfunction depends on the type of abnormality seen. Recurrent infections due to reflux are treated with appropriate antibiotics. If structural changes have been identified then twice-yearly urinalysis and work-up for functional abnormality should be done. A few patients would require dialysis or even renal transplant. Testosterone supplements have not been beneficial for patients with genital abnormalities.11 Puberty is a particularly stressful time for these patients and counseling can be of immense help. Control of diabetes can prevent diabetic retinopathy and rapid deterioration of vision already compromised by rod-cone dystrophy. A cardiologist should be consulted
for suspected cardiovascular problem. An orthopedic surgeon may be consulted for possible
excision of excess digits to help the patient maximize function. Specially fitted shoes are beneficial to patients with bone deformation. A dietician or nutritionist should be engaged in addressing obesity, which is a major source of stress for both patient and parents. Given the complex etiology of weight gain in Bardet-Biedl, a multidisciplinary approach is recommended in dealing with weight loss. This may include a combination of careful dietary assessment, diet, behavioral therapy, and exercise. Learning difficulties should be addressed early, before visual impairment interferes, if possible. Special schools can aid in providing a program for these patients. We have described a patient with Bardet-Biedl syndrome and is the first reported case at the Philippine General Hospital.
1. Beales PL, Elcioglu N, Woolf AS, et al. New criteria for improved diagnosis of BardetBiedl syndrome: results of a population survery. J Med Genet 1999; 36: 437-446.
2. Mykytyn K, Nishimura DY, Searby CC, Shastri M., et al Identification of the gene (BBS1) most commonly involved in Bardet-Biedl syndrome, a complex human
obesity syndrome. Nat Genet 2002; 31: 435-438.
3. Online Mendelian Inheritance in Man (OMIM™). Johns Hopkins University. Bardet Biedl Syndrome. MIM number (209900). April 4, 2003: http://www.ncbi.nlm.nih.gov/ omim (Accessed April 28,2003).
4. Farag TI,Teebi AS. High incidence of Bardet-Biedl syndrome among the Bedouins. Clin Genet 1989; 36: 463-465.
5. Millay RH, Weleber RG, Heckenlively JR. Ophthalmologic and systemic manifestations of Alstrom’s disease. Am J Ophthalmol 1986; 102: 482-490.
6. Schachat A, Maumenee IH. Bardet-Biedl syndrome and related disorders. Arch Ophthalmol 1982; 100: 285-288.
7. Ryan SJ. Retina, 3rd ed. Vol 1. St. Louis, Missouri: Mosby, 2001; 423-425.
8. Iannaccone A, De Propris G, Roncati S, et al. The ocular phenotype of the BardetBiedl syndrome. Comparison to nonsyndromic retinitis pigmentosa. Ophthalmic Genet 1997; 18: 13-26.
9. Green JS, Parfrey PS, Jarnett JD, et al. The cardinal manifestations of BardetBiedl syndrome, a form of Laurence-Moon-Biedl syndrome. N Engl J Med 1989; 321: 1002-1009.
10. Harnett JD, Green JS, Cramer BC, et al. The spectrum of renal disease in LaurenceMoon-Biedl syndrome. N Engl J Med 1988; 319: 615-618.
11. Campo RV, Aaberg TM. Ocular and systemic manifestations of Bardet-Biedl Syndrome. Am J Ophthalmol 1982; 94: 751-756.